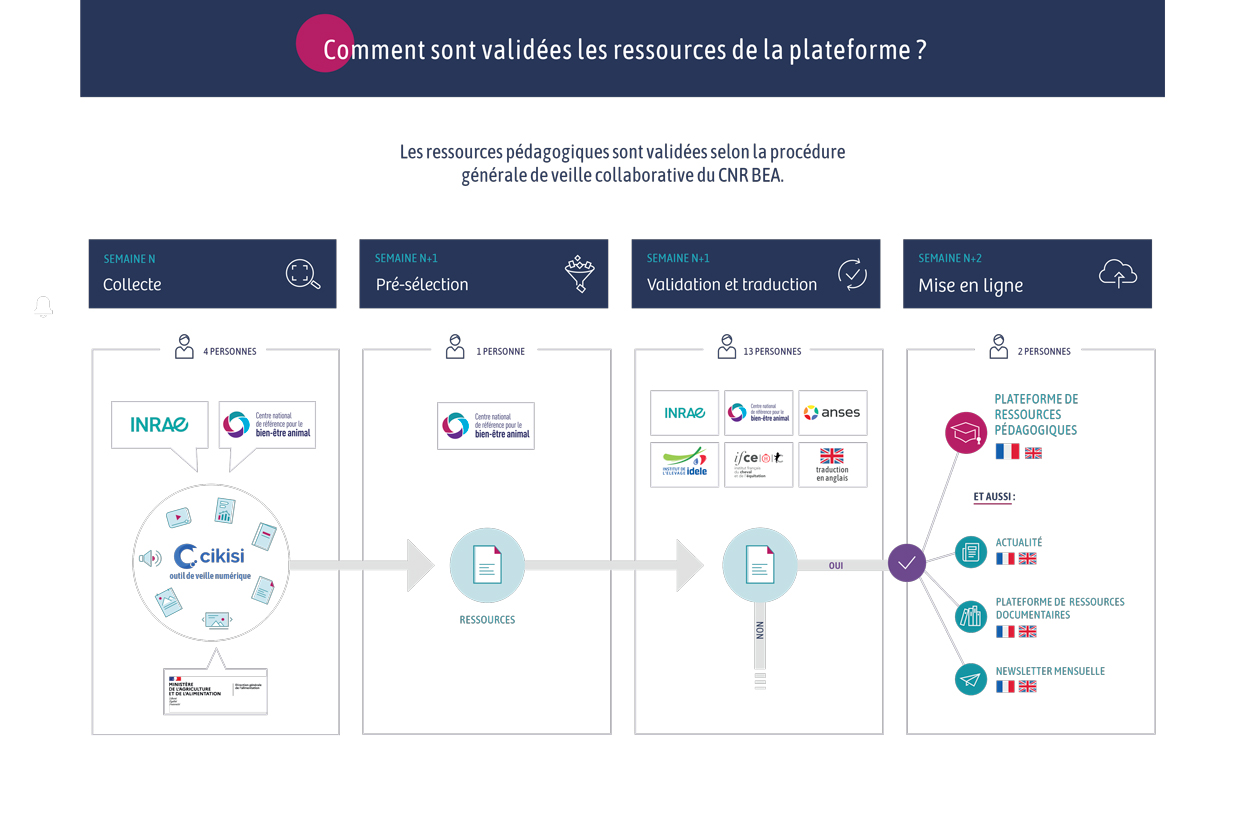
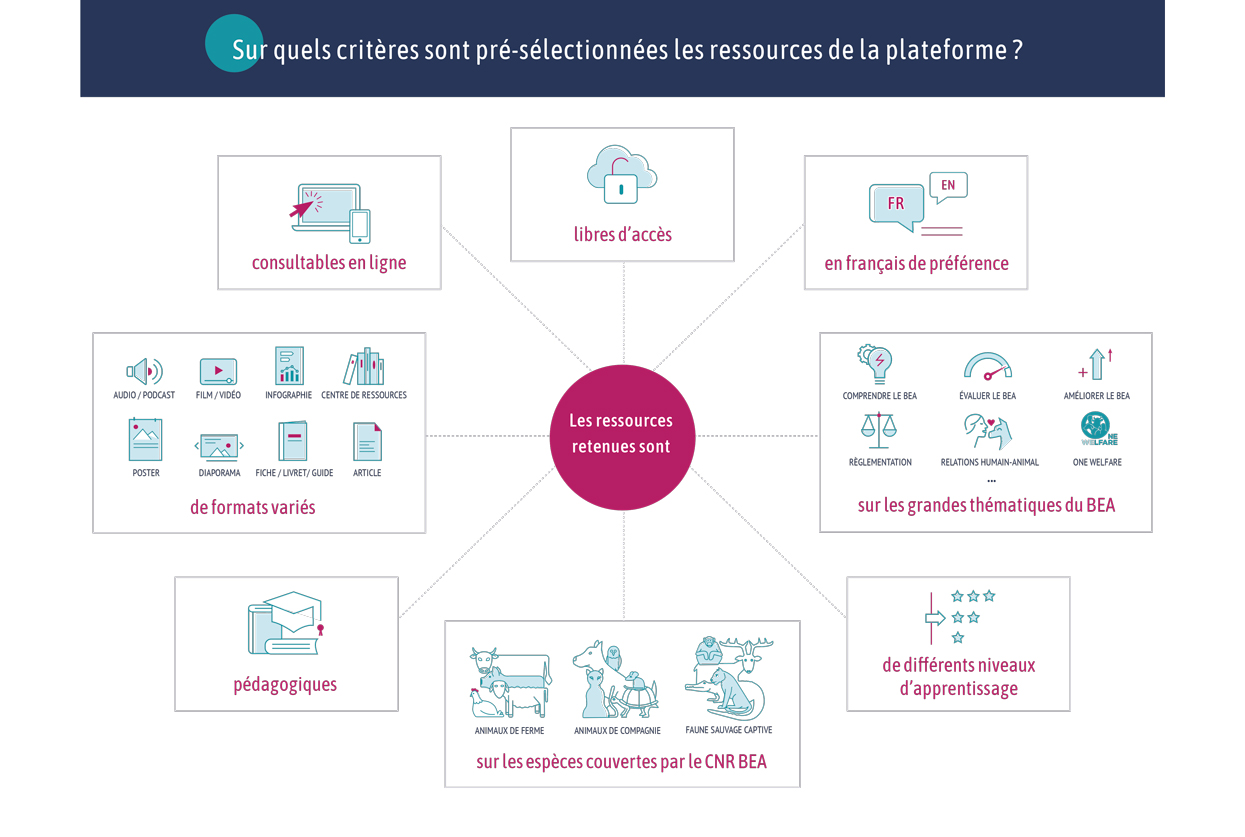
Type de document : article publié dans Frontiers in Veterinary Science
Auteurs : Fábio Pértille, Adriana Mercia Guaratini Ibelli, Maj El Sharif, Mirele Daiana Poleti, Anna Sophie Fröhlich, Shiva Rezaei, Mônica Corrêa Ledur, Per Jensen, Carlos Guerrero-Bosagna, Luiz Lehmann Coutinho
Résumé en français (traduction) : Les animaux de production sont constamment soumis à des conditions environnementales défavorables à un stade précoce qui influencent le phénotype adulte et produisent des effets épigénétiques. La méthylation des dinucléotides CpG dans les globules rouges (GR) pourrait être un biomarqueur épigénétique utile pour identifier les animaux soumis à un stress chronique dans l’environnement de production. Nous avons comparé ici une fraction réduite du méthylome des GR de poulets exposés à l’isolement social à des poulets non exposés. Ces expériences ont été réalisées dans deux pays différents : le Brésil (BR) et la Suède (SW). L’objectif était d’identifier les profils de méthylation de l’ADN associés au stress dans les GR de ces populations, malgré les conditions variables auxquelles les oiseaux sont exposés dans chaque installation et les différentes lignées. Les oiseaux étaient de plus en plus exposés à un traitement d’isolement social, combiné à une privation de nourriture et d’eau, à des périodes aléatoires de la journée, de la première à la quatrième semaine après l’éclosion. Nous avons ensuite recueilli l’ADN des GR des individus et comparé une fraction réduite de leur méthylome entre les groupes expérimentaux en utilisant deux approches bioinformatiques pour identifier les régions différemment méthylées (RDM) : une utilisant des fenêtres de taille fixe et une autre qui présélectionne les pics différentiels avec le MACS2. Trois niveaux de signification ont été utilisés (P ≤ 0.05, P ≤ 0.005, et P ≤ 0.0005) pour identifier les RDM entre les groupes expérimentaux, qui ont ensuite été utilisés pour différentes analyses. Avec les deux approches, un plus grand nombre de RDM ont atteint les seuils de significativité définis chez les individus du BR par rapport à ceux de SW. Toutefois, un plus grand nombre de RDM présentaient des valeurs de changement plus élevées dans le groupe SW par rapport aux individus du groupe BR. Il est intéressant de noter que ChrZ a été enrichi au-delà des attentes en ce qui concerne la présence de RDM. De plus, en analysant l’emplacement de ces RDM par rapport au site de départ de la transcription (SDT), nous avons trouvé trois pics avec une forte présence de RDM: 10 kb en amont, le SDT lui-même, et 20-40 kb en aval. Il est intéressant de noter que ces pics présentaient des RDM avec une forte proportion (>50%) de sites de liaison de facteurs de transcription spécifiques. Trois RDM se chevauchant ont été trouvées entre la population BR et SW en utilisant la p-value la plus détendue (P ≤ 0,05). Avec la valeur p la plus stricte (P ≤ 0,0005), nous avons trouvé 7 et 4 DMR entre les traitements dans les populations BR et SW, respectivement. Cette étude est la première approximation permettant d’identifier les biomarqueurs épigénétiques de l’exposition à long terme au stress dans différentes lignées d’animaux de production
Résumé en anglais (original) : Production animals are constantly subjected to early adverse environmental conditions that influence the adult phenotype and produce epigenetic effects. CpG dinucleotide methylation in red blood cells (RBC) could be a useful epigenetic biomarker to identify animals subjected to chronic stress in the production environment. Here we compared a reduced fraction of the RBC methylome of chickens exposed to social isolation to non-exposed. These experiments were performed in two different locations: Brazil and Sweden. The aim was to identify stress-associated DNA methylation profiles in RBC across these populations, in spite of the variable conditions to which birds are exposed in each facility and their different lineages. Birds were increasingly exposed to a social isolation treatment, combined with food and water deprivation, at random periods of the day from weeks 1–4 after hatching. We then collected the RBC DNA from individuals and compared a reduced fraction of their methylome between the experimental groups using two bioinformatic approaches to identify differentially methylated regions (DMRs): one using fixed-size windows and another that preselected differential peaks with MACS2. Three levels of significance were used (P ≤ 0.05, P ≤ 0.005, and P ≤ 0.0005) to identify DMRs between experimental groups, which were then used for different analyses. With both of the approaches more DMRs reached the defined significance thresholds in BR individuals compared to SW. However, more DMRs had higher fold change values in SW compared to BR individuals. Interestingly, ChrZ was enriched above expectancy for the presence of DMRs. Additionally, when analyzing the locations of these DMRs in relation to the transcription starting site (TSS), we found three peaks with high DMR presence: 10 kb upstream, the TSS itself, and 20–40 kb downstream. Interestingly, these peaks had DMRs with a high presence (>50%) of specific transcription factor binding sites. Three overlapping DMRs were found between the BR and SW population using the most relaxed p-value (P ≤ 0.05). With the most stringent p-value (P ≤ 0.0005), we found 7 and 4 DMRs between treatments in the BR and SW populations, respectively. This study is the first approximation to identify epigenetic biomarkers of long-term exposure to stress in different lineages of production animals.
Publication ayant fait l’objet d’un article sur le site Lab Worldwide : Biomarkers Reveal Stress Level in Chickens